
SAM/BAM quality control: Analyzing short read quality (after mapping)
Contents
- Remove Duplicates
- Determine the paired-end insert size for DNA samples
- QC to get a (visual) summary of mapping statistics
- Graphically analyze read duplication
- Interpret quality control issues
Remove Duplicates
Remove duplicates, for example, when one molecule is amplified via PCR and sequenced multiple times.
# Use 'samtools rmdup' # on single-end reads (-s option) or paired-end reads (-S option) samtools rmdup [-sS] <input.srt.bam> <output.bam>
Determine the paired-end insert size for DNA samples
If paired-end insert size or distance is unknown or need to be verified, it can be extracted from a BAM/SAM file after running an unspliced mapper.
When mapping with bowtie (or another mapper), the insert size can often be included as an input parameter (example for bowtie: -X 500), which can help with mapping. See our mapping SOP for mapping details.
Method 1: Get insert sizes from BAM file
# Using a SAM file (at Unix command prompt)
awk -F "\t" '$9 > 0 {print $9}' s_1_bowtie.sam > s_1_insert_sizes.txt
# Using a BAM file (at Unix command prompt)
samtools view s_1_bowtie.bam | awk -F"\t" '$9 > 0 {print $9}' > s_1_insert_sizes.txt
# and then process column of numbers with R (or Excel)
# In R Session
sizeFile = "s_1_insert_sizes.txt"
sample.name = "My paired reads"
distance = read.delim(sizeFile, h=F)[,1]
pdf(paste(sample.name, "insert.size.histogram.pdf", sep="."), w=11, h=8.5)
hist(distance, breaks=200, col="wheat", main=paste("Insert sizes for", sample.name), xlab="length (nt)")
dev.off()
Method 2: Calculate insert sizes with CollectInsertSizeMetrics function from picard. This is also a good approximation for RNA samples.
# # I=File Input SAM or BAM file. (Required) # O=File File to write the output to. (Required) # H=File File to write insert size histogram chart to. (Required) # output: CollectInsertSizeMetrics.txt: values for -r and --mate-std-dev can be found in this text file # CollectInsertSizeMetrics_hist.pdf: insert size histogram (graphic representation) sbatch --partition=20 --job-name=picard_insert --mem=8G --wrap "java -jar /usr/local/share/picard-tools/picard.jar CollectInsertSizeMetrics I=foo.bam O=CollectInsertSizeMetrics.txt H=CollectInsertSizeMetrics_hist.pdf"
You might need to specify a different java path if above command is not working. On local tak, you can use /usr/local/jre1.8/bin/java
QC to get a (visual) summary of mapping statistics
This includes the coverage/distribution of mapped reads across the genome or transcriptome
Use Picard CollectRnaSeqMetrics.jar to find coverage across gene body for 5' or 3' bias
[RNA-seq only] Get global coverage profile across transcripts
Do reads come from across the length of a typical transcript, or is there 3' or 5' bias (where most reads come from one end of a typical transcript)?
One way to look at this is with Picard's CollectRnaSeqMetrics tool
# Usage: java -jar picard.jar CollectRnaSeqMetrics INPUT=bamFile REF_FLAT=refFlatFile STRAND_SPECIFICITY=NONE OUTPUT=outputFile CHART_OUTPUT=output.pdf VALIDATION_STRINGENCY=SILENT # Example command java -jar /usr/local/share/picard-tools/picard.jar CollectRnaSeqMetrics INPUT=WT.bam REF_FLAT=/nfs/genomes/mouse_mm10_dec_11_no_random/anno/refFlat.txt STRAND_SPECIFICITY=NONE OUTPUT=QC_metrics/WT.RnaSeqMetrics.txt CHART_OUTPUT=QC_metrics/WT.RnaSeqMetrics.pdf VALIDATION_STRINGENCY=SILENT
The VALIDATION_STRINGENCY=SILENT option will keep the program from crashing if it finds something unexpected. The default: VALIDATION_STRINGENCY=STRICT
QualiMap can be used on DNA or RNA-Seq to get summary of mapping and coverage/distribution
# For Graphical interface (with GUI access to all tools) qualimap # Full command on the command line: # Before submitting to cluster unset DISPLAY sbatch --partition=20 --job-name=qualimap_bamqc --mem=8G --wrap "qualimap bamqc -bam myFile.bam -outdir output_qualimap" # For huge data, you can increase memory with --java-mem-size="4800M" to avoid OutOfMemoryError: Java heap space # For rnaseq QC sbatch --partition=20 --job-name=qualimap_rnaseqqc --mem=8G --wrap "qualimap rnaseq -bam myFile.bam -gtf Homo_sapiens.GRCh37.72.canonical.gtf -outdir output_qualimap_rnaseq -p non-strand-specific" # For counts QC (after using htseq-count or a similar program to generate a matrix of counts) qualimap counts -d countsqc_input.txt -c -s HUMAN -outdir counts_qc #Format of countsqc_input.txt (below), totalCounts.txt is a matrix of counts; header lines must be commented "#" and species is human or mouse only. #Sample Condition Path Column HMLE1 HMLE totalCounts.txt 2 HMLE2 HMLE totalCounts.txt 3 HMLE3 HMLE totalCounts.txt 4 N81 N8 totalCounts.txt 5 N82 N8 totalCounts.txt 6 N83 N8 totalCounts.txt 7
RSeQC is a RNA-Seq quality control package for getting mapping statistics (eg. unique/multi-mapped reads)
bam_stat.py -i myFile.bam
# Or run on a folder of BAMs
for bamFile in `/bin/ls *.bam`; do sbatch --partition=20 --job-name=bamstat_${bamFile%.bam} --mem=4G --wrap "bam_stat.py -i $bamFile > $bamFile.bam_stat.txt"; done
Use infer_experiment.py from the RseQC package to check if/how your RNA-seq reads are stranded.
# Command line:
sbatch --partition=20 --job-name=infer_exp --mem=4G --wrap "infer_experiment.py -i My_sample.accepted_hits.bam -r my_genes.bed > My_sample.infer_experiment.out.txt"
-i INPUT_FILE in SAM or BAM format
-r Reference gene models in bed format (converted from GTF file).
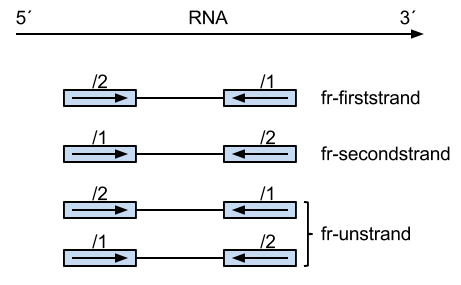
--library-type=fr-unstranded
--library-type=fr-firststrand
--library-type=fr-secondstrand
# sample output on strand-specific PE reads (since the first fraction is much larger than the second fraction):
This is PairEnd Data
Fraction of reads explained by "1++,1--,2+-,2-+": 0.9807
Fraction of reads explained by "1+-,1-+,2++,2--": 0.0193
Fraction of reads explained by other combinations: 0.0000
# For gene counting: (featureCounts, use -p -s 1; htseq-count, use --stranded=yes); mapping with TopHat should have been performed with --library-type=fr-secondstrand.
# sample output on strand-specific PE reads (since the second fraction is much larger than the first fraction):
This is PairEnd Data
Fraction of reads explained by "1++,1--,2+-,2-+": 0.0193
Fraction of reads explained by "1+-,1-+,2++,2--": 0.9807
Fraction of reads explained by other combinations: 0.0000
# For gene counting: (featureCounts, use -p -s 2; htseq-count, use --stranded=reverse); mapping with TopHat should have been performed with --library-type=fr-firststrand.
# sample output on non-stranded PE reads (since both fractions are about the same):
This is PairEnd Data
Fraction of reads explained by "1++,1--,2+-,2-+": 0.5103
Fraction of reads explained by "1+-,1-+,2++,2--": 0.4897
Fraction of reads explained by other combinations: 0.0000
# For gene counting: (featureCounts, use -p -s 0; htseq-count, use --stranded=no); mapping with TopHat should have been performed with --library-type=fr-unstranded.
#sample output on stranded SE reads:
This is SingleEnd Data
Fraction of reads failed to determine: 0.0068
Fraction of reads explained by "++,--": 0.9865
Fraction of reads explained by "+-,-+": 0.0068
# For gene counting: (featureCounts, use -s 1; htseq-count, use --stranded=yes; mapping with TopHat should have been performed with --library-type=fr-secondstrand.
#sample output on stranded SE reads:
This is SingleEnd Data
Fraction of reads failed to determine: 0.0068
Fraction of reads explained by "++,--": 0.0068
Fraction of reads explained by "+-,-+": 0.9865
# For gene counting: (featureCounts, use -s 2; htseq-count, use --stranded=reverse; mapping with TopHat should have been performed with --library-type=fr-firststrand.
For paired-end RNA-seq, there are two different ways to strand reads:
i) 1++,1--,2+-,2-+
read1 mapped to '+' strand indicates parental gene on '+' strand
read1 mapped to '-' strand indicates parental gene on '-' strand
read2 mapped to '+' strand indicates parental gene on '-' strand
read2 mapped to '-' strand indicates parental gene on '+' strand
ii) 1+-,1-+,2++,2--
read1 mapped to '+' strand indicates parental gene on '-' strand
read1 mapped to '-' strand indicates parental gene on '+' strand
read2 mapped to '+' strand indicates parental gene on '+' strand
read2 mapped to '-' strand indicates parental gene on '-' strand
For single-end RNA-seq, there are two different ways to strand reads:
i) ++,--
read mapped to '+' strand indicates parental gene on '+' strand
read mapped to '-' strand indicates parental gene on '-' strand
ii) +-,-+
read mapped to '+' strand indicates parental gene on '-' strand
read mapped to '-' strand indicates parental gene on '+' strand
Figure from Tophat/Bowtie library options

PE Reads Orientation
Most PE reads (from Illumina) should be FR (--> <--), others include FF (--> --> or <-- <--), or RF (<-- -->) and these might be from structural variation.
Graphically analyze read duplication
The R/Bioconductor package dupRadar can do this, analyzing a BAM file that has had duplicates flagged (such as with Picard's MarkDuplicates tool).
A set of commands can be run with an R script by the package authors available from their Using the dupRadar package page.
A BaRC script (/nfs/BaRC_Public/BaRC_code/R/dupRadar/dupRadar.R) does both the duplicate marking and the analysis with a command like
# Usage: ./dupRadar.R <file.bam> <genes.gtf> <stranded=[no|yes|reverse]> paired=[yes|no] outdir=./ threads=1 sbatch --partition=20 --job-name=dupRadar --mem=8G --wrap "/nfs/BaRC_Public/BaRC_code/R/dupRadar/dupRadar.R WT.bam /nfs/genomes/mouse_mm10_dec_11_no_random/gtf/Mus_musculus.GRCm38.81.canonical.gtf stranded=no paired=yes outdir=dupRadar_out threads=1"
Interpret quality control issues
See QCFAIL.com from the Babraham Institute
Attachments (1)
- tophat_library.png (11.8 KB ) - added by 10 years ago.
{kind=link}
Download all attachments as: .zip