
Quality Control and preprocessing of short reads
FASTQ:
Format
Each entry in a FASTQ file consists of four lines:
- Sequence identifier
- Sequence
- Quality score identifier line (consisting of a +)
- Quality score
Naming
- Ensure your fastq headers conforms to the standarad convention listed below, otherwise, downstream analysis (eg. aligners, counting, etc.) may behave differently than intended.
FASTQ header/naming scheme as specified by Illumina's CASAVA pipeline,
Illumina (Casava 1.8.2):
@<instrument>:<run number>:<flowcell ID>:<lane>:<tile>:<x-pos>:<y-pos> <read>:<is filtered>:<control number>:<index sequence>
| Element | Requirements | Description |
| @ | @ | Each sequence identifier line starts with @ |
| <instrument> | Characters allowed: a-z, A-Z, 0-9 and underscore | Instrument ID (eg. HWI-DXXXX for HiSeq 2500) |
| <run number> | Numerical | Run number on instrument |
| <flowcell ID> | Characters allowed: a-z, A-Z, 0-9 | |
| <lane> | Numerical | Lane number |
| <tile> | Numerical | Tile number |
| <x_pos> | Numerical | X coordinate of cluster |
| <y_pos> | Numerical | Y coordinate of cluster |
| <read> | Numerical | Read number. 1 can be single read or read 2 of paired-end |
| <is filtered> | Y or N | Y if the read is filtered, N otherwise |
| <control number> | Numerical | 0 when none of the control bits are on, otherwise it is an even number |
| <index sequence> | ACTG | Index sequence |
Illumina (Casava 1.7)
@<machine_id>:<lane>:<tile>:<x_coord>:<y_coord>#<index>/<read_#>
| Element | Requirements | Description |
| @ | @ | Each sequence identifier line starts with @ |
| <machine_id> | Characters allowed: a-z, A-Z, 0-9 and underscore | Instrument ID (eg. HWI-DXXXX for HiSeq 2500) |
| <lane> | Numerical | Lane number |
| <tile> | Numerical | Tile number |
| <x_coord> | Numerical | X coordinate of cluster |
| <y_coord> | Numerical | Y coordinate of cluster |
| #<index> | 0 or 1 | 0 means no index, 1 means indexed sample |
| /<read_#> | Numerical | Read number. 1 can be single read or read 2 of paired-end |
Analyzing short read quality (before mapping)
Quality scoring introduction
- Quality scores are typically represented using a Phred scoring scheme, where a read quality value = −10 * log10 (error probability)
- For example,
- Quality = 10 => error rate = 10% => base call has 90% confidence
- Quality = 20 => error rate = 1% => base call has 99% confidence
- Quality = 30 => error rate = 0.1% => base call has 99.9% confidence
- See Phred quality score for more details.

FastQC
- quality control analysis with nice graphical output
- available for Linux, Windows, and MacOSX
- (but no tools for editing reads)
It's installed on fry and can be run from the command line
- Sample command 1 (fastq inputs): fastqc s_1_sequence.txt s_2_sequence.txt
- Sample command 2 (fastq.gz inputs): fastqc s_1_sequence.txt.gz s_2_sequence.txt.gz
or interactively (with X Windows):
- fastqc
Output is a directory (named "s_1_sequence_fastqc" for input "s_1_sequence.txt") with "fastqc_report.html", a web page including all figures.
The "Basic Statistics" section at the top of the FastQC report also shows the Encoding (quality score) information (like "Illumina 1.5"), which may be necessary to specify in subsequent analysis steps. The Encoding scales are described at http://en.wikipedia.org/wiki/FASTQ_format#Encoding.
MultiQC
MultiQC is a program that can merge QC and other reports (such as FastQC, STAR, and MACS2) for a series of samples into one big file. See MultiQC: Supported Tools for a table of about 100 types of reports that can be merged.
The main command to merge reports in your current directory is
# Usage: multiqc [OPTIONS] <analysis directory> multiqc -n My_samples.reports.html .
Note the " .", which indicates the current directory. Otherwise, a series of directories can be used as arguments.
QC for paired-end reads
- QC as above each of the forward and reverse reads separately using QC'ing program (above).
- If reads reads are removed, get reads/mates after QC'ing that are perfect pairs:
sbatch --partition=20 --job-name=cmpfastq --mem=16G --wrap "/nfs/BaRC_Public/BaRC_code/Perl/cmpfastq/cmpfastq.pl s_8_1_sequence.txt s_8_2_sequence.txt" # fastq inputs
ShortRead (R package)
ShortRead
R package, Linux (Tak), Window, Mac
- It takes the fastq files, and creates a website with different ways to check the data, and with instruction on how to interpret results. The output files are stored in dest folder (my_qa in this example).
- QC a single file using ShortRead
library("ShortRead")
# load the data
sr <- readFastq("s1_sequence.txt")
# create a qa object from the ShortRead object
qa <- qa( sr, lane="character" )
# create an html report in the qa directory
report(qa, dest="my_qa")
- QC all *.txt fastq files in a directory using ShortRead.
library("ShortRead")
qaSummary <- qa(".", pattern="*.txt", type="fastq")
#create an html report in the qa directory
report(qaSummary, dest="myQC_dir")
Fastx Toolkit
FastX Toolkit
galaxy integration, Linux(Tak), MacOSX
Note: fastq_quality_filter v 0.0.14 may have a bug where it reports "got empty array at fastq_quality_filter.c:97" and/or segmentation fault.
# Sample commands: # quality_stats: Sample Solexa reads file: s_1_1_sequence.txt or s_1_1_sequence.txt.gz fastx_quality_stats -i s_1_1_sequence.txt -o s_1_1_sequence.stats # fastq input gunzip -c s_1_1_sequence.txt.gz | fastx_quality_stats -o s_1_1_sequence.stats # fastq.gz input # a figure for Nucleotide Distribution: fastx_nucleotide_distribution_graph.sh -i s_1_1_sequence.stats -o s_1_1_sequence.stats.nuc.png -t "s_1_1_sequence.stats Nucleotide Distribution" # boxplot: fastq_quality_boxplot_graph.sh -i s_1_1_sequence.stats -o s_1_1_sequence.stats.quality.png -t "s_1_1_sequence.stats Quality Scores"
PrinSeq
PrinSeq gives summary statistics and QC info, eg. exact duplicate and other options, includes visual summary on a fastq file.
#Get html summary of output prinseq-lite.pl -fastq myFile.fastq -graph_data file.gd -out_good null -out_bad null prinseq-graphs.pl -i file.gd -html_all -o prinseq_outFile #remove exact duplicates (drep=1) and create a new fastq file (of unique sequences) prinseq-lite.pl -fastq myFile.fastq -derep 1 -out_good myFile.unique.fq -out_bad myFile.dup.fastq
Modifying a file of short reads based on quality considerations
Remove reads with low quality score: To use FASTX Toolkit to get only reads that are above a quality score along with a certain read length:
fastq_quality_filter -v -q 20 -p 75 -i myFile.fastq -o myFile.fastq.fastx_trim # fastq input and output gunzip -c myFile.fq.gz | fastq_quality_filter -v -q 20 -p 75 -z -o myFile.fastq.fastx_trim.gz # fastq.gz input and output [-h] = This helpful help screen. [-q N] = Minimum quality score to keep. [-p N] = Minimum percent of bases that must have [-q] quality. [-z] = Compress output with GZIP. [-i INFILE] = FASTA/Q input file. default is STDIN. [-o OUTFILE] = FASTA/Q output file. default is STDOUT. [-v] = Verbose - report number of sequences. If [-o] is specified, report will be printed to STDOUT. If [-o] is not specified (and output goes to STDOUT), report will be printed to STDERR.
If you get an error like "Invalid quality score value" or segmentation fault, your fastq file probably has Sanger (offset 33) instead of Illumina (ASCII offset 64) quality scores. You'll need to add the option "-Q33" to your FASTX Toolkit arguments.
Trim end of reads when quality drops below a threshold
- sample command:
sbatch --partition=20 --job-name=trim --mem=16G --wrap "fastq_quality_trimmer -v -t 20 -l 25 -i input.fastq -o output.fastq" # fastq input and output
sbatch --partition=20 --job-name=trim --mem=16G --wrap "gunzip -c input.fastq.gz | fastq_quality_trimmer -v -t 20 -l 25 -z -o output.fastq.gz" # fastq.gz input and output
[-t N] = Quality threshold - nucleotides with lower
quality will be trimmed (from the end of the sequence).
[-l N] = Minimum length - sequences shorter than this (after trimming)
will be discarded. Default = 0 = no minimum length.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTQ input file. default is STDIN.
[-o OUTFILE] = FASTQ output file. default is STDOUT.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
Modifying a file of short reads in other ways
Remove linker (adapter) RNA:
- What is the sequence of the linker (adapter) to be removed?
- Biologists generally know which linker (adapter) RNA is used for their sample(s).
- For experiments using Illumina libraries, What sequences do I use for adapter trimming? may help you find the correct adapter sequence(s). The above adapters may be used with KAPA kits as well.
- Also or in addition, when you run quality control with shortRead or FASTQC, check out
- repetitive segments in the "over represented sequences" section.
- "Per base sequence content" for any patterns at the beginning of your reads
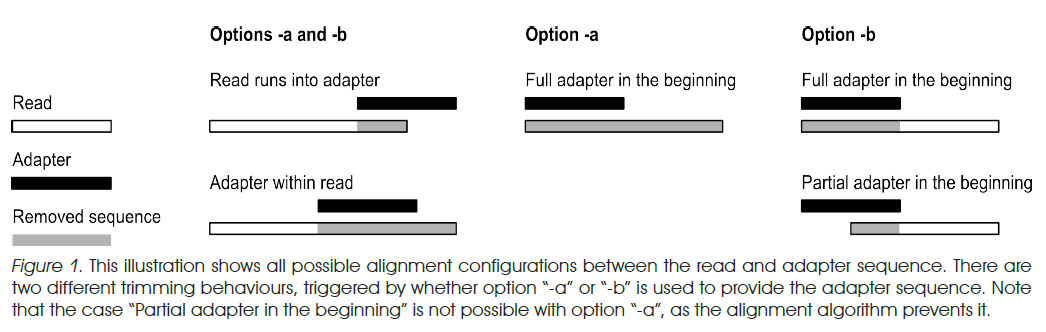
- Method 1: cutadapt is a good tool that is designed to find and remove adapters:
- more options than fastx_clipper, such as specifically trimming 5' or 3' adapters and specifying error rate (allowed mismatches)
- much more conservative than fastx_clipper.
- In paired-end mode, if a read is removed from one file, its pair will be removed from the other file
- guide
- a vs b options from EMBnet.journal

- sample commands:
# single-end reads sbatch --partition=20 --job-name=cutadapt --mem=16G --wrap "cutadapt -a GATCGGAAGAGCTCGTATGCCGTCTT -o Reads_noAdapter.fastq Reads.fastq" In the above command: -a: Sequence of an adapter that was ligated to the 3' end. -o: output file name # paired-end reads sbatch --partition=20 --job-name=cutdapt --mem=16G --wrap "cutadapt -a GATCGGAAGAGCTCGTATGCCGTCTT -o Reads_trimmed.1.fq -p Reads_trimmed.2.fq Reads.1.fq Reads.2.fq" # use --quality-base=64 option for Phred+64 (Illumina 1.5)
- To remove Illumina TruSeq adapters (from the cutadapt user guide):
# Choose your preferred minimum length. If this is not set, some output reads may have a length of 0, which can cause downstream applications to fail. cutadapt --minimum-length 25 -a AGATCGGAAGAGC -A AGATCGGAAGAGC -o trimmed.1.fq.gz -p trimmed.2.fq.gz reads.1.fq.gz reads.2.fq.gz
- Method 2: Trim Galore! combines cutadapt, FastQC, and the ability to guess what your adapters, if -a/-a2 options are not provided:
- Sample command
# Usage (single-end reads): trim_galore [options] <filename(s)> trim_galore --phred64 --fastqc -o my_trimmed_reads raw_reads/My_reads.fq.gz # Usage (paired-end reads; discard pair if at least one read is too short): trim_galore --paired --phred64 --fastqc -o my_trimmed_reads raw_reads/My_reads_1.fq.gz raw_reads/My_reads_2.fq.gz # Usage (paired-end reads, reduced-representation bisulfite sequencing (RRBS) data, default phred33 quality scores): trim_galore --paired --rrbs --fastqc -o my_trimmed_reads raw_reads/My_reads_1.fq.gz raw_reads/My_reads_2.fq.gz # Get all options trim_galore --help --phred64 ==> Use Illumina 1.5 encoding quality scores --fastqc ==> Run FastQC after trimming -o ==> Print output files in this directory instead of the current directory
- Sample command
- Method 3: fastx_clipper
- Sample command:
sbatch --partition=20 --job-name=clip --mem=16G --wrap "fastx_clipper -a CTGTAGGCACCATCAAT -i s2_sequence.txt -v -l 22 -o s2_sequence_noLinker.txt" # fastq input and output sbatch --partition=20 --job-name=clip --mem=16G --wrap "gunzip -c s2_sequence.txt | fastx_clipper -a CTGTAGGCACCATCAAT -v -l 22 -z -o s2_sequence_noLinker.txt.gz" # fastq.gz input and output In the above command: -a CTGTAGGCACCATCAAT is the linker sequence -i s2_sequence.txt is input solexa fastq file -v is Verbose [report number of sequences in output and discarded] -l 22 is to discard sequences shorter than 22 nucleotides -o s2_ sequence_noLinker.txt is output file.
- Sample command:
- See fastx_clipper usage (or fastx_clipper -h) for more arguments
- If you get the message "Invalid quality score value..." you have the older range of quality scores.
- Add the argument -Q 33, such as
- fastx_clipper -a CTGTAGGCACCATCAAT -Q 33 -i s2_sequence.txt -v -l 22 -o s2_sequence_noLinker.txt
Trim reads to a specified length
- If we have reads of different lengths (i.e. because we clipped out the adapter sequences), we can trim them to have them all be the same length. Use fastx_trimmer for that.
- sample command:
sbatch --partition=20 --job-name=trim --mem=16G --wrap "fastx_trimmer -f 1 -l 22 -i s7_sequences.fastq -o s7_sequences_trimmed.fastq" # fastq input and output sbatch --partition=20 --job-name=trim --mem=16G --wrap "gunzip -c s7_sequences.fastq.gz | fastx_trimmer -f 1 -l 22 -z -o s7_sequences_trimmed.fastq.gz" # fastq.gz input and output [-i INFILE] = FASTA/Q input file. default is STDIN. [-o OUTFILE] = FASTA/Q output file. default is STDOUT. [-l N] = Last base to keep [-f N] = First base to keep. Default is 1 (=first base).
Select reads that are paired [for paired-end sequencing]
During quality control, if low-quality reads have been removed for any reason, some reads may not have a paired end at the other end. This can cause problems with mapping programs.
- Method 1:
Sample command:
/nfs/BaRC_Public/BaRC_code/Perl/cmpfastq/cmpfastq.pl sequence.1_1.filt.txt sequence.1_2.filt.txt # fastq inputs
Output files will be
- *unique.out (reads that are only in the "1" or "2" set; 2 files) and
- *common.out (reads that are in both the "1" and "2" set; 2 files).
The *common.out reads should be used for paired-read mapping.
- Method 2:
For fastq.gz files use the seqkit instead seqkit
seqkit pair -1 sequence.1_1.filt.fq.gz -2 sequence.1_2.fil.fq.gz # fastq inputs
Select a subset of reads by read ID
To get a list of read IDs from a fastq file, one can use a command like
zmore Reads.fq.gz | awk 'NR % 4 == 1' | perl -pe 's/^\@//' >| Selected_read_IDs.txt
Using this list of read IDs, one can extract this subset of reads from a larger fastq file (using seqtk) like
seqtk subseq All_reads.fq.gz Selected_read_IDs.txt >| Selected_reads.fq
Analyzing potential species composition/contamination of reads in a fastq file
- To get a quick preview of what genomes/collections (or vectors or other contaminants) of sequences the reads in a fastq file can map to, one can use the FastQ Screen tool.
- Information about FastQ Screen can be found at this page: http://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/
- FastQ Screen allows you to screen your fastq file against a set of libraries, which can be set up to contain the genomes of interest, potential contaminating genomes, vectors, sequencing adaptors, ribosomal RNAs, or other contaminants commonly seen in sequencing experiments. This allows you to see if composition of the reads matches with what you expect and, where the contamination might have come from, if any.
- FastQ Screen uses bowtie or bowtie2 for mapping. The libraries you wish to screen against need to be bowtie-indexed.
- Paths to the bowtie indexed libraries need to be specified in the fastq_screen.conf file, which is called either from the same directory where the fastq_screen program is (by default) or from a manually specified location using the -conf /path/to/.conf file option
- Sample commands are:
Usage: fastq_screen [OPTIONS] file.fq eg. fastq_screen --aligner bowtie2 myFastQ.txt Commonly used options are: [--aligner] Specify 'bowtie' or bowtie2' to use for the mapping. [--outdir] Specify a directory in which to save output files. [--illumina1_3] Assume that the quality values are in encoded in Illumina v1.3 format. Defaults to Sanger format if this flag is not specified. [--conf] Manually specify a location for the configuration file to be used for this run. If not specified then the file will be taken from the same directory as the fastq_screen program. Note: the config file is already setup on our internal server
Analyzing short read quality (after mapping)
Attachments (3)
- cutadapt.jpeg (17.5 KB ) - added by 12 years ago.
- cutadapt.2.jpeg (109.1 KB ) - added by 12 years ago.
- fastq_phread-base.png (68.6 KB ) - added by 10 years ago.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Download all attachments as: .zip